Parcourir les catégories
Explorer
Fiverr Pro
Français
$
USD
Passionné par l'apprentissage
À propos de ce service
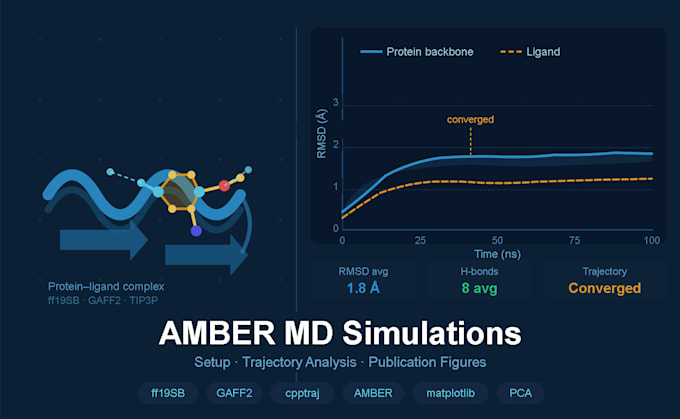
Travaillez-vous sur un système protéine-ligand et avez-vous besoin de simulations de dynamique moléculaire fiables et bien analysées pour votre recherche ou publication ? Je propose des services complets de simulation MD avec AMBER, de la préparation du système à la réalisation de figures prêtes pour publication en utilisant le même pipeline que celui utilisé dans la recherche en découverte de médicaments computationnelle évaluée par des pairs.
Je suis un chercheur en biologie computationnelle avec une expérience pratique dans AMBER/AmberTools, cpptraj, et les workflows d’analyse basés sur Python. Mon travail a directement soutenu des manuscrits soumis à des revues à comité de lecture en pharmacologie computationnelle.
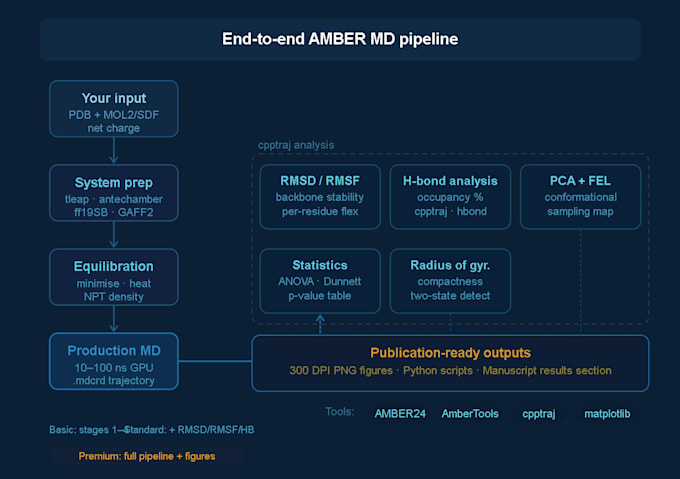
Ce que je fournis
Configuration du système
Préparation de la structure de la protéine et attribution de l’état de protonation
Paramétrisation du ligand avec antechamber
Solvatation (bassin d’eau TIP3P ou OPC), ajout d’ions de contre-ions
Affectation du champ de force : ff19SB (protéine), GAFF2 (petite molécule)
Minimisation, chauffage et équilibrage NPT/NVT
Exécution de la MD de production
Trajectoires de 10, 50 ou 100 ns (dépendant du package)
Fichiers de trajectoire .mdcrd complets fournis
Analyse de la trajectoire (Standard & Premium)
Figures prêtes pour publication complète
Pourquoi travailler avec moi ?
Une vraie expérience amber, pas un simple wrapper cloud ou un outil en ligne.