Parcourir les catégories
Explorer
Fiverr Pro
Français
$
USD
Expert en chimioinformatique et RDKit
Je vais traiter et visualiser vos données chimiques en utilisant Python et RDKit rapidement et avec précision, pour la recherche de médicaments ou la recherche scientifique.
Ce que je peux faire :
- Convertir SMILES/SDF/CSV en rapports Excel
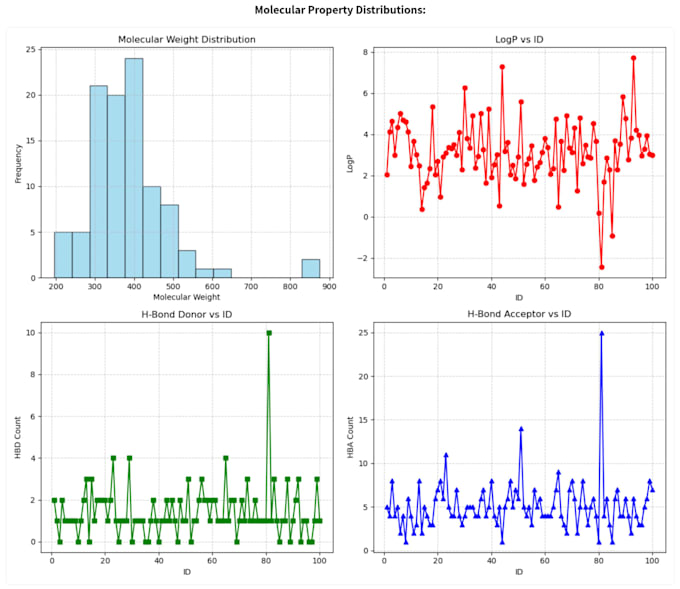
- Calculer les principaux descripteurs (Masse molaire, LogP, TPSA, HBA, HBD)
- Appliquer les filtres de la règle des cinq de Lipinski
- Générer des empreintes et effectuer des recherches de similarité
- Créer des cartes de chaleur de similarité et des diagrammes de diversité chimique
- Construire des notebooks Jupyter réutilisables avec tout le code
Pourquoi me choisir ?
- Master en chimie organique (Université de Shanghai + SIOC, CAS)
- 2 articles SCI sur la réaction de Heck asymétrique
- Plus de 4 ans d’expérience dans la sélection de blocs de construction similaires aux médicaments
- Python (RDKit, Pandas, scikit-learn)
Différents packages pour divers volumes de molécules. Contactez-moi pour des besoins spécifiques.
Ce dont j’ai besoin de votre part :
- Fichier moléculaire (SMILES, CSV ou SDF)
- Brève description de l’objectif
Commandez dès maintenant ou contactez-moi !



Domaine:
Machine learning
•
Autres
Expertise:
Classification
•
Analyse prédictive
Langage de programmation:
Python
Outils:
Jupyter Notebook
•
Excel
•
Autres
Technologie:
Python
•
scikit-learn
•
Jupyter Notebook
•
Pandas
•
Excel
Traduction automatique
Quels formats de fichiers pouvez-vous accepter ?
Je peux accepter une liste de SMILES (CSV/Excel/TXT), des fichiers SDF ou tout fichier texte avec une colonne de SMILES. Indiquez simplement le nom de la colonne si ce n’est pas standard.
Pouvez-vous traiter un grand nombre de molécules ?
Oui, mon processus est optimisé pour jusqu’à 5000 molécules dans le forfait Premium. Pour des bibliothèques plus importantes, veuillez me contacter d’abord pour un devis personnalisé.
Ai-je besoin de connaître Python ou RDKit pour utiliser votre service ?
Pas du tout. Vous fournissez simplement votre fichier moléculaire et me dites quelle analyse vous souhaitez. Je livrerai les résultats sous Excel ou sous forme d’un notebook Jupyter prêt à l’emploi – aucun codage requis de votre côté.
Pouvez-vous personnaliser l’analyse selon les besoins spécifiques de mon projet, même au-delà de la chimformatique standard ?
Absolument. Si vous avez un jeu de données unique (par exemple, données de réaction, spectroscopie ou autres tableaux chimiques/biologiques) ou si vous avez besoin d’une visualisation spécifique ou d’un script Python personnalisé, envoyez-moi simplement les détails. Je suis heureux de proposer une solution adaptée – pas limitée aux descripteurs moléculaires standards.
Offrez-vous un support continu ou des modifications après livraison ?
Oui, je propose des révisions raisonnables dans le cadre de la commande initiale. Si vous avez besoin de modifications supplémentaires, d’un support étendu ou d’aide pour adapter le code à un autre jeu de données, nous pouvons discuter d’un petit supplément. Mon objectif est de m’assurer que vous soyez entièrement satisfait du résultat final.